在聚合物科学中,我们常把材料性能首先归因于化学结构,比如单体是什么、链之间怎么连接、分子有没有极性、相互作用强不强等。但越来越多的研究表明,就算化学组成完全一样,只要把分子的“形状和连接方式”换一换,也就是改变它的拓扑结构,材料的表现就可能发生明显变化。比如同样的链,做成环、星形、瓶刷,或者再把它们交联成网络,都会带来宏观上性能的显著改变。
在此背景下,机械互锁分子与机械互锁聚合物(mechanically interlocked molecules/polymers, MIMs/MIPs)提供了一类尤为独特的拓扑平台。与传统聚合物依赖共价键连接不同,机械互锁体系的组分通过“机械键”被拓扑锁在一起:它们能够相对运动(例如滑动、旋转),却无法在不发生共价断裂的情况下分离。这种“可动但不散”的连接方式,使得材料在受力与松弛过程中具备额外的自由度与额外的耗能通道,也因此被寄予在韧性增强、应力重分配、可编程响应与分子机器等方向上的潜力。然而,机械互锁体系的一个长期困难在于:宏观性能的差异来自于介观尺度的运动学过程,例如环沿轴的滑移动力学、索烃之间的相对转动、受限空间中的结在拉伸下的局域收紧等。许多过程发生在实验难以直接解析的尺度区间,且往往由多个变量耦合控制。正因如此,分子模拟尤其是粗粒化分子动力学成为理解这类材料的重要入口。
近期,颜徐州团队在Macromolecules以“Progress in Molecular Dynamics Simulations of Mechanically Interlocked Polymers”为题发表Perspective文章,系统梳理了机械互锁和相关聚合物的分子动力学研究进展,并强调了多尺度建模在该领域的必要性与方法学要点。
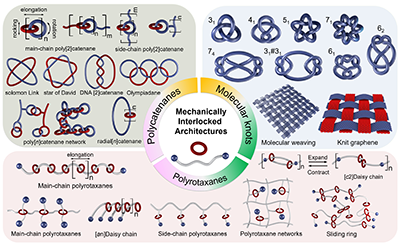
图1. 机械互锁分子和相关的聚合物、网络的结构多样性示意图,分为聚链烷、聚轮烷和分子结。
该综述以粗粒化分子动力学(Coarse-Grained Molecular Dynamics,CGMD)为重点,梳理了机械互锁分子与聚合物近年来的模拟研究进展,从最基本的轮烷、索烃和分子结,到结构更复杂的线性机械互锁聚合物与机械互锁网络,展示了模拟如何揭示“滑动、穿线与识别位点”等微观运动对力传递、能量耗散与松弛行为的决定作用,如图1所示。
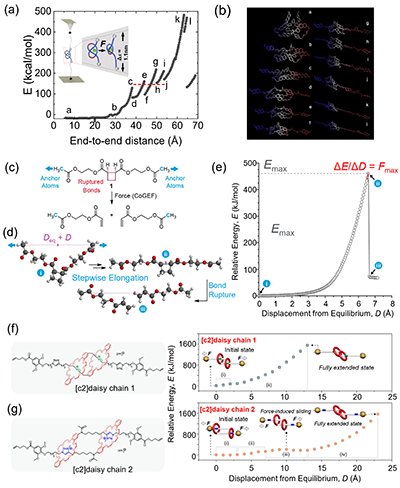
图2. 量子化学(Quantum chemistry,QC)计算和约束几何模拟外力(constrained geometries simulate external force,COGEF)方法。
同时,该综述还将不同分辨率的模拟方法放回它们最适合回答的问题框架中。量子化学与CoGEF等方法可以在最高分辨率上处理相互作用能垒、外力作用下的反应路径以及化学键断裂阈值等问题,为后续模型提供标定依据,如图2所示。CoGEF(Constrained Geometries Simulate External Force)是一种在量子化学框架下“近似模拟外力拉伸”的方法,它通过对分子施加端到端距离约束并逐步增大该约束,得到体系能量随拉伸位移变化的曲线,从而间接推导出等效的力–位移响应与关键阈值。它的优势在于实现简单、计算成本相对可控,能够直观捕捉受力过程中构型如何演化、哪个结构单元最先被激活以及断裂或反应发生前后的结构差异,因此常用于比较不同分子设计在机械加载下的相对稳定性、预测可能的受力敏感位点,并为后续全原子或粗粒化模型的势函数选择与参数标定提供参考。
全原子分子动力学(All-atom Molecular Dynamics,AAMD)的核心优势在于“把细节保留下来”,如图3所示,它显式表示每个原子(通常包含氢原子),并把水、离子等环境因素直接纳入体系,因此能够真实呈现氢键网络的形成与破裂、溶剂化壳层的重排、离子在带电基团或识别位点附近的富集与屏蔽效应。这些微观相互作用会直接改写互锁体系的自由能景观,例如轮烷环在轴上的驻留位置、在不同识别位点之间切换的能垒、以及滑动过程中受到的摩擦与阻滞。借助自由能计算(如自适应偏置力等方法),AAMD 可以把“看起来在动”的构象变化定量化为能垒高度与稳定态分布,从而解释实验中观察到的开关行为、响应速率和环境敏感性。受限于计算尺度,它更适合单个互锁单元或短链体系的机制解析与参数标定。
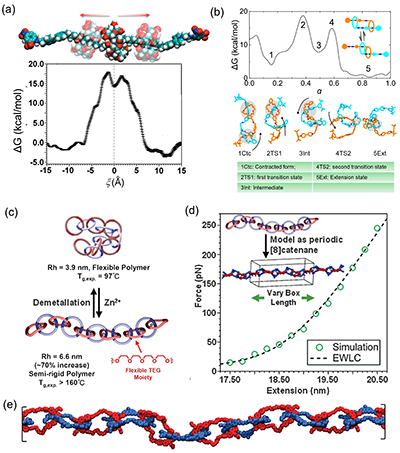
图3. 机械互锁分子和机械互锁聚合物的全原子分子动力学模拟。
当关注点从化学细节转向拓扑决定的普适规律时,粗粒化分子动力学更具优势,如图4所示。它用更简化的粒子表示体系,显著降低计算成本,使模拟能够覆盖更大的时间与空间尺度,从而直接观察环的滑移、链的穿线、网络在拉伸下的重排与断裂等介观过程,并把这些微观运动与模量、韧性、耗能和松弛等宏观性质建立起可定量的联系。
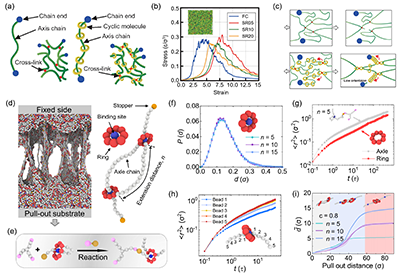
图4. 机械互锁聚合物网络的力学、动力学和环滑动特性。
在文章结尾,作者给出了对机械互锁材料研究的展望:未来真正重要的并不只是把模拟“算得更大、更久”,而是把方法做得更规范、更可复用,并且能和实验形成更可靠的验证闭环。一个核心挑战是,在粗粒化层级如何既保留拓扑主导的物理本质,又能合理地引入识别位点、金属配位、离子特异效应等化学因素,让模型既“抓住关键”,又不过度简化。另一个难点来自网络体系本身的复杂性,真实互锁网络往往存在交联分散、缺陷、环密度不均,以及拓扑纠缠与互锁约束相互耦合等问题,因此需要更标准化的结构构建与表征流程,才能让不同研究之间可对比、可复现。作者还强调“机械互锁vitrimer”是很有潜力的方向:动态共价交换提供可重构网络,机械键提供可滑移的拓扑自由度,两者可能在应力松弛、韧化与加工性之间形成新的协同。要把这种协同从概念变成可用的设计规律,离不开模拟与实验的相互印证。总体来说,以拓扑为核心变量的分子动力学,正在把互锁材料的微观运动转化为可计算、可比较、能反推设计的规律,为这一领域从“能合成”走向“可设计”提供关键支撑。
图文转载自【中国聚合物网】:http://www.polymer.cn/